
T-cell receptor
| TCR complex | |
|---|---|
 | |
| Identifiers | |
| Symbol | TCR |
| OPM superfamily | 166 |
| Membranome | 26 |

| T-cell receptor alpha locus | |
|---|---|
| Identifiers | |
| Symbol | TRA |
| Alt. symbols | TCRA, TRA@ |
| NCBI gene | 6955 |
| HGNC | 12027 |
| OMIM | 186880 |
| Other data | |
| Locus | Chr. 14 q11.2 |
| T-cell receptor beta locus | |
|---|---|
| Identifiers | |
| Symbol | TRB |
| Alt. symbols | TCRB, TRB@ |
| NCBI gene | 6957 |
| HGNC | 12155 |
| OMIM | 186930 |
| Other data | |
| Locus | Chr. 7 q34 |
| T-cell receptor delta locus | |
|---|---|
| Identifiers | |
| Symbol | TRD |
| Alt. symbols | TCRD, TRD@, TCRDV1 |
| NCBI gene | 6964 |
| HGNC | 12252 |
| Other data | |
| Locus | Chr. 14 q11.2 |
| T-cell receptor gamma locus | |
|---|---|
| Identifiers | |
| Symbol | TRG |
| Alt. symbols | TCRG, TRG@ |
| NCBI gene | 6965 |
| HGNC | 12271 |
| Other data | |
| Locus | Chr. 7 p14 |
The T-cell receptor (TCR) is a protein complex found on the surface of T cells, or T lymphocytes,[1] that is responsible for recognizing fragments of antigen as peptides bound to major histocompatibility complex (MHC) molecules. The binding between TCR and antigen peptides is of relatively low affinity and is degenerate: that is, many TCRs recognize the same antigen peptide and many antigen peptides are recognized by the same TCR.[2]
The TCR is composed of two different protein chains (that is, it is a heterodimer). In humans, in 95% of T cells the TCR consists of an alpha (α) chain and a beta (β) chain (encoded by TRA and TRB, respectively), whereas in 5% of T cells the TCR consists of gamma and delta (γ/δ) chains (encoded by TRG and TRD, respectively). This ratio changes during ontogeny and in diseased states (such as leukemia). It also differs between species. Orthologues of the 4 loci have been mapped in various species.[3][4] Each locus can produce a variety of polypeptides with constant and variable regions.[3]
When the TCR engages with antigenic peptide and MHC (peptide/MHC), the T lymphocyte is activated through signal transduction, that is, a series of biochemical events mediated by associated enzymes, co-receptors, specialized adaptor molecules, and activated or released transcription factors. Based on the initial receptor triggering mechanism, the TCR belongs to the family of non-catalytic tyrosine-phosphorylated receptors (NTRs).[5]
History
[edit]In 1982, Nobel laureate James P. Allison first discovered a clonally expressed T-cell surface epitope in murine T lymphoma.[6] In 1983, Ellis Reinherz first defined the structure of the human T-cell receptor using anti-idiotypic monoclonal antibodies to T-cell clones, complemented by studies in the mouse by Philippa Marrack and John Kappler.[7][8] Then, Tak Wah Mak[9] and Mark M. Davis[10] identified the cDNA clones encoding the human and mouse TCR respectively in 1984. These findings allowed the entity and structure of the elusive TCR, known before as the "Holy Grail of Immunology", to be revealed. This allowed scientists from around the world to carry out studies on the TCR, leading to important studies in the fields of CAR-T, cancer immunotherapy and checkpoint inhibition.
Structural characteristics
[edit]The TCR is a disulfide-linked membrane-anchored heterodimeric protein normally consisting of the highly variable alpha (α) and beta (β) chains expressed as part of a complex with the invariant CD3 chain molecules. T cells expressing this receptor are referred to as α:β (or αβ) T cells, though a minority of T cells express an alternate receptor, formed by variable gamma (γ) and delta (δ) chains, referred as γδ T cells.[11]
Each chain is composed of two extracellular domains: Variable (V) region and a Constant (C) region, both of Immunoglobulin superfamily (IgSF) domain forming antiparallel β-sheets. The Constant region is proximal to the cell membrane, followed by a transmembrane region and a short cytoplasmic tail, while the Variable region binds to the peptide/MHC complex.
The variable domain of both the TCR α-chain and β-chain each have three hypervariable or complementarity-determining regions (CDRs). There is also an additional area of hypervariability on the β-chain (HV4) that does not normally contact antigen and, therefore, is not considered a CDR.[citation needed]
The residues in these variable domains are located in two regions of the TCR, at the interface of the α- and β-chains and in the β-chain framework region that is thought to be in proximity to the CD3 signal-transduction complex.[12] CDR3 is the main CDR responsible for recognizing processed antigen, although CDR1 of the alpha chain has also been shown to interact with the N-terminal part of the antigenic peptide, whereas CDR1 of the β-chain interacts with the C-terminal part of the peptide[citation needed].
CDR2 is thought to recognize the MHC. HV4 of the β-chain is not thought to participate in antigen recognition as in classical CDRs, but has been shown to interact with superantigens.[13]
The constant domain of the TCR consists of short connecting sequences in which a cysteine residue forms disulfide bonds, which form a link between the two chains.
The TCR is a member of the immunoglobulin superfamily, a large group of proteins involved in binding, recognition, and adhesion; the family is named after antibodies (also called immunoglobulins). The TCR is similar to a half-antibody consisting of a single heavy and single light chain, except the heavy chain is without its crystallizable fraction (Fc). The two main subunits of TCR (α- and β-chains) are twisted together. CD3 and zeta subunits are required to carry out the signal transduction. The MHC-TCR-CD3 interaction for T cells is functionally similar to the antigen(Ag)-immunoglobulin(Ig)-FcR interaction for myeloid leukocytes, and Ag-Ig-CD79 interaction for B cells.
Generation of the TCR diversity
[edit]The generation of TCR diversity is similar to that for antibodies and B-cell antigen receptors. It arises mainly from genetic recombination of the DNA-encoded segments in individual somatic T cells by somatic V(D)J recombination using RAG1 and RAG2 recombinases. Unlike immunoglobulins, however, TCR genes do not undergo somatic hypermutation, and T cells do not express activation-induced cytidine deaminase (AID). The recombination process that creates diversity in BCR (antibodies) and TCR is unique to lymphocytes (T and B cells) during the early stages of their development in primary lymphoid organs (thymus for T cells, bone marrow for B cells).
Each recombined TCR possess unique antigen specificity, determined by the structure of the antigen-binding site formed by the α and β chains in case of αβ T cells or γ and δ chains on case of γδ T cells.[14]
- The TCR alpha chain is generated by VJ recombination, whereas the beta chain is generated by VDJ recombination (both involving a random joining of gene segments to generate the complete TCR chain).
- Likewise, generation of the TCR gamma chain involves VJ recombination, whereas generation of the TCR delta chain occurs by VDJ recombination.
The intersection of these specific regions (V and J for the alpha or gamma chain; V, D, and J for the beta or delta chain) corresponds to the CDR3 region that is important for peptide/MHC recognition (see above).
It is the unique combination of the segments at this region, along with palindromic and random nucleotide additions (respectively termed "P-" and "N-"), which accounts for the even greater diversity of T-cell receptor specificity for processed antigenic peptides.
Later during development, individual CDR loops of TCR can be re-edited in the periphery outside thymus by reactivation of recombinases using a process termed TCR revision (editing) and change its antigenic specificity.
The TCR complex
[edit]In the plasma membrane the TCR receptor chains α and β associate with six additional adaptor proteins to form an octameric complex. The complex contains both α and β chains, forming the ligand-binding site, and the signaling modules CD3δ, CD3γ, CD3ε and CD3ζ in the stoichiometry TCR α β - CD3εγ - CD3εδ - CD3ζζ. Charged residues in the transmembrane domain of each subunit form polar interactions allowing a correct and stable assembly of the complex.[15] The cytoplasmic tail of the TCR is very short, hence the CD3 adaptor proteins containing the signaling motifs are needed for propagating the signal from the triggered TCR into the cell.
The signaling motifs involved in TCR signaling are tyrosine residues in the cytoplasmic tail of these adaptor proteins that can be phosphorylated in the event of TCR-pMHC binding. The tyrosine residues reside in a specific amino acid sequence of the signature Yxx(L/I)x6-8Yxx(L/I), where Y, L, I indicate tyrosine, leucine and isoleucine residues, x denotes any amino acids, the subscript 6-8 indicates a sequence of 6 to 8 amino acids in length. This motif is very common in activator receptors of the non-catalytic tyrosine-phosphorylated receptor (NTR) family and is referred to as immunoreceptor tyrosine-based activation motif (ITAM).[5] CD3δ, CD3γ and CD3ε each contain a single ITAM, while CD3ζ contains three ITAMs. In total the TCR complex contains 10 ITAMs.[15] Phosphorylated ITAMs act as binding site for SH2-domains of additionally recruited proteins.
Antigen discrimination
[edit]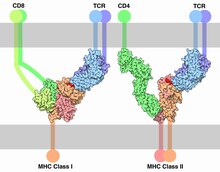
Each T cell expresses clonal TCRs which recognize a specific peptide loaded on a MHC molecule (pMHC), either on MHC class II on the surface of antigen-presenting cells or MHC class I on any other cell type.[16] A unique feature of T cells is their ability to discriminate between peptides derived from healthy, endogenous cells and peptides from foreign or abnormal (e.g. infected or cancerous) cells in the body.[17] Antigen-presenting cells do not discriminate between self and foreign peptides and typically express a large number of self-derived pMHCs on their cell surface and only a few copies of any foreign pMHC. For example, cells infected with HIV have only 8–46 HIV-specific pMHCs, compared with 100,000 total pMHCs, per cell.[18][19]
Because T cells undergo positive selection in the thymus, there is a non-negligible affinity between self-pMHC and the TCR. Nevertheless, the T-cell receptor signaling should not be activated by self-pMHC so that endogenous, healthy cells are ignored by T cells. However, when these very same cells contain even minute quantities of pathogen-derived pMHC, T cells must get activated and initiate immune responses. The ability of T cells to ignore healthy cells but respond when these same cells express a small number of foreign pMHCs is known as antigen discrimination.[20][21]
To do so, T cells have a very high degree of antigen specificity, despite the fact that the affinity to the peptide/MHC ligand is rather low in comparison to other receptor types.[22] The affinity, given as the dissociation constant (Kd), between a TCR and a pMHC was determined by surface plasmon resonance (SPR) to be in the range of 1–100 μM, with an association rate (kon) of 1000 -10000 M−1 s−1 and a dissociation rate (koff) of 0.01 -0.1 s−1.[23] In comparison, cytokines have an affinity of KD = 10–600 pM to their receptor.[24] It has been shown that even a single amino acid change in the presented peptide that affects the affinity of the pMHC to the TCR reduces the T-cell response and cannot be compensated by a higher pMHC concentration.[25] A negative correlation between the dissociation rate of the pMHC-TCR complex and the strength of the T-cell response has been observed.[26] That means, pMHC that bind the TCR for a longer time initiate a stronger activation of the T cell. Furthermore, T cells are highly sensitive; interaction with a single pMHC is enough to trigger activation.[27] T cells move on quickly from antigens that do not trigger responses, rapidly scanning pMHC on an antigen-presenting cell (APC) to increase the chance of finding a specific pMHC. On average, a T cell encounters 20 APCs per hour.[28]
Different models for the molecular mechanisms that underlie this highly specific and highly sensitive process of antigen discrimination have been proposed. The occupational model simply suggests that the TCR response is proportional to the number of pMHC bound to the receptor. Given this model, a shorter lifetime of a peptide can be compensated by higher concentration such that the maximum response of the T cell stays the same. However, this cannot be seen in experiments and the model has been widely rejected.[26] The most accepted view is that the TCR engages in kinetic proofreading. The kinetic proofreading model proposes that a signal is not directly produced upon binding but a series of intermediate steps ensure a time delay between binding and signal output. Such intermediate "proofreading" steps can be multiple rounds of tyrosine phosphorylation. These steps require energy and therefore do not happen spontaneously, only when the receptor is bound to its ligand. This way only ligands with high affinity that bind the TCR for a long enough time can initiate a signal. All intermediate steps are reversible, such that upon ligand dissociation the receptor reverts to its original unphosphorylated state before a new ligand binds.[29] This model predicts that maximum response of T cells decreases for pMHC with shorter lifetime. Experiments have confirmed this model.[26] However, the basic kinetic proofreading model has a trade-off between sensitivity and specificity. Increasing the number of proofreading steps increases the specificity but lowers the sensitivity of the receptor. The model is therefore not sufficient to explain the high sensitivity and specificity of TCRs that have been observed. (Altan Bonnet2005) Multiple models that extend the kinetic proofreading model have been proposed, but evidence for the models is still controversial.[17][30][31]
The antigen sensitivity is higher in antigen-experienced T cells than in naive T cells. Naive T cells pass through the process of functional avidity maturation with no change in affinity. It is based on the fact that effector and memory (antigen-experienced) T cell are less dependent on costimulatory signals and higher antigen concentration than naive T cell.[32]
Signaling pathway
[edit]The essential function of the TCR complex is to identify specific bound antigen derived from a potentially harmful pathogen and elicit a distinct and critical response. At the same time it has to ignore any self-antigen and tolerate harmless antigens such as food antigens. The signal transduction mechanism by which a T cell elicits this response upon contact with its unique antigen is termed T-cell activation. Upon binding to pMHC, the TCR initiates a signaling cascade, involving transcription factor activation and cytoskeletal remodeling resulting in T-cell activation. Active T cells secrete cytokines, undergo rapid proliferation, have cytotoxic activity and differentiate into effector and memory cells. When the TCR is triggered, T cells form an immunological synapse allowing them to stay in contact with the antigen presenting cell for several hours.[33] On a population level, T-cell activation depends on the strength of TCR stimulation, the dose–response curve of ligand to cytokine production is sigmoidal. However, T-cell activation on a single cell level can be characterized by a digital switch-like response, meaning the T cell is fully activated if the stimulus is higher than a given threshold; otherwise the T cell stays in its non-activated state. There is no intermediate activation state. The robust sigmoid dose-response curve on population level results from individual T cells having slightly different thresholds.[25]
T cells need three signals to become fully activated. Signal 1 is provided by the T-cell receptor when recognising a specific antigen on a MHC molecule. Signal 2 comes from co-stimulatory receptors on T cell such as CD28, triggered via ligands presented on the surface of other immune cells such as CD80 and CD86. These co-stimulatory receptors are expressed only when an infection or inflammatory stimulus is detected by the innate immune system, Known as a "Danger signal". This two-signal system makes sure that T cells only respond to harmful stimuli (i.e. pathogens or injury) and not to self-antigens. An additional third signal is provided by cytokines, which regulate the differentiation of T cells into different subsets of effector T cells.[33] There are a myriad of molecules involved in the complex biochemical process (called trans-membrane signaling) by which T-cell activation occurs. Below, the signaling cascade is described in detail.
Receptor activation
[edit]The initial triggering follows the mechanism common for all NTR receptor family members. Once the TCR binds a specific pMHC, the tyrosine residues of the immunoreceptor tyrosine-based activation motifs (ITAMs) in its CD3 adaptor proteins are phosphorylated. The residues serve as docking sites for downstream signaling molecules, which can propagate the signal.[34][35] Phosphorylation of ITAMs is mediated by the Src kinase Lck. Lck is anchored to the plasma membrane by associating with the co-receptor CD4 or CD8, depending on the T-cell subtype. CD4 is expressed on helper T cells and regulatory T cells, and is specific for MHC class II. CD8, on the other hand, specific for MHC class I, is expressed on cytotoxic T cells. Binding of the co-receptor to the MHC brings Lck in close proximity to the CD3 ITAMs. It has been shown that 40% of Lck is active even before the TCR binds pMHC and therefore has the ability to constantly phosphorylate the TCR.[36] Tonic TCR signalling is avoided by the presence of phosphatase CD45 that removes phosphorylation from tyrosine residues and inhibits signal initiation. Upon binding the balance of kinase activity to phosphatase activity is perturbed, leading to a surplus of phosphorylation and initiation of the signal. How such perturbation is accomplished by TCR binding is still debated. Mechanisms involving conformational change of TCR, TCR aggregation and kinetic segregation have been suggested.[34] Tyrosine kinase Fyn might be involved in ITAM phosphorylation but is not essential for TCR signaling.[37][38]
Proximal TCR signaling
[edit]Phosphorylated ITAMs in the cytoplasmic tails of CD3 recruit protein tyrosine kinase Zap70 that can bind to the phosphorylated tyrosine residues with its SH2 domain. This brings Zap70 into close proximity to Lck which results to its phosphorylation and activation by Lck.[39] Lck phosphorylates a number of different proteins in the TCR pathway.[40] Once activated, Zap70 is able to phosphorylate multiple tyrosine residues of the transmembrane protein LAT. LAT is a scaffold protein associated with the membrane. It itself does not have any catalytic activity but it provides binding sites for signalling molecules via phosphorylated tyrosine residues. LAT associates with another scaffolding protein Slp-76 via the Grap2 adaptor protein, which provides additional binding sites. Together LAT and Slp-76 provide a platform for the recruitment of many downstream signaling molecules. By bringing these signalling molecules into close proximity, they can then be activated by Lck, Zap70 and other kinases. Therefore, the LAT/Slp76 complex act as a highly cooperative signalosome.[39]
Molecules that bind the LAT/Slp76 complex include: Phospholipase Cγ1 (PLCγ1), SOS via a Grb2 adaptor, Itk, Vav, Nck1 and Fyb.[39]
Signal transduction to the nucleus
[edit]PLCγ is a very important enzyme in the pathway as it generates second messenger molecules. It is activated by the tyrosine kinase Itk which is recruited to the cell membrane by binding to phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3 is produced by the action of phosphoinositide 3-kinase(PI-3K), which phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to produce PIP3. It is not known that PI-3K is activated by the T-cell receptor itself, but there is evidence that CD28, a co-stimulatory receptor providing the second signal, is able to activate PI-3K. The interaction between PLCγ, Itk and PI-3K could be the point in the pathway where the first and the second signal are integrated. Only if both signals are present, PLCγ is activated.[33] Once PLCγ is activated by phosphorylation. It hydrolyses PIP2 into two secondary messenger molecules, namely the membrane-bound diacyl glycerol (DAG) and the soluble inositol 1,4,5-trisphosphate (IP3).[41]
These second messenger molecules amplify the TCR signal and distribute the prior localized activation to the entire cell and activate protein cascades that finally lead to the activation of transcription factors. Transcription factors involved in T-cell signaling pathway are the NFAT, NF-κB and AP1, a heterodimer of proteins Fos and Jun. All three transcription factors are needed to activate the transcription of interleukin-2(IL2) gene.[33]
NFAT
[edit]NFAT activation depends on calcium signaling. IP3 produced by PLC-γ is no longer bound to the membrane and diffuses rapidly in the cell. Binding of IP3 to calcium channel receptors on the endoplasmic reticulum (ER) induces the release of calcium (Ca2+) into the cytosol. The resulting low Ca2+ concentration in the ER causes STIM1 clustering on the ER membrane, which in turn leads to activation of cell membrane CRAC channels that allows additional calcium to flow into the cytosol from the extracellular space. Therefore, levels of Ca2+ are strongly increased in the T cell. This cytosolic calcium binds calmodulin, inducing a conformational change of the protein such that it can then bind and activate calcineurin. Calcineurin, in turn, dephosphorylates NFAT. In its deactivated state, NFAT cannot enter the nucleus as its nuclear localization sequence (NLS) cannot be recognized by nuclear transporters due to phosphorylation by GSK-3. When dephosphorylated by Calcineurin translocation of NFAT into the nucleus is possible.[33] Additionally, there is evidence that PI-3K via signal molecules recruits the protein kinase AKT to the cell membrane. AKT is able to deactivate GSK3 and thereby inhibiting the phosphorylation of NFAT, which could contribute to NFAT activation.[39]
NF-κB
[edit]NF-κB activation is initiated by DAG, the second, membrane bound product of PLCγ hydrolyzation of PIP2. DAG binds and recruits protein kinase C θ (PKCθ) to the membrane where it can activate the membrane bound scaffold protein CARMA1. CARMA1 then undergoes a conformational change which allows it to oligomerize and bind the adapter proteins BCL10, CARD domain and MALT1. This multi-subunit complex binds the ubiquitin ligase TRAF6. Ubiquitination of TRAF6 serves as scaffold to recruit NEMO, IκB kinase (IKK) and TAK1.[33] TAK 1 phosphorylates IKK, which in turn phosphorylates the NF-κB inhibitor I-κB, leading to the ubiquitination and subsequent degradation of I-κB. I-κB blocks the NLS of NF-κB therefore preventing its translocation to the nucleus. Once I-κB is degraded, it cannot bind to NF-κB and the NLS of NF-κB becomes accessible for nuclear translocation.[33]
AP1
[edit]Activation of AP1 factor involves three MAPK signaling pathways. These pathways use a phosphorylation cascade of three successive acting protein kinases to transmit a signal. The three MAPK pathways in T cells involve kinases of different specificities belonging to each of the MAP3K, MAP2K, MAPK families. Initial activation is done by the GTPase Ras or Rac which phosphorylate the MAP3K.[33] A cascade involving the enzymes Raf, MEK1, ERK results in the phosphorylation of Jun, conformational change allows Jun to bind to Fos and hence AP-1 to form. AP-1 then acts as transcription factor. Raf is activated via the second messenger DAG, SOS, and Ras. DAG recruits among other proteins the RAS guanyl nucleotide-releasing protein (RasGRP), a guanine nucleotide exchange factor (GEF), to the membrane. RasGRP activates the small GTPase Ras by exchanging guanosine diphosphate (GDP) bound to Ras against guanosine triphosphate (GTP). Ras can also be activated by the guanine nucleotide exchange factor SOS which binds to the LAT signalosome. Ras then initiates the MAPK cascade.[39] The second MAPK cascade with MEKK1, JNKK, JNK induces protein expression of Jun. Another cascade, also involving MEKK1 as MAPK3, but then activating MKK3 /6 and p38 induces Fos transcription. Activation of MEKK1, additionally to being activated by Ras, involves Slp-76 recruiting the GEF Vav to the LAT signalosome, which then activates the GTPase Rac. Rac and Ras activate MEKK1 and thereby initiate the MAPK cascade.[39]
See also
[edit]References
[edit]- ^ Kindt TJ, Goldsby RA, Osborne BA, Kuby J (2007). Kuby immunology. Macmillan. pp. 223–. ISBN 978-1-4292-0211-4. Retrieved 28 November 2010.
- ^ Sewell AK (September 2012). "Why must T cells be cross-reactive?". Nature Reviews. Immunology. 12 (9): 669–77. doi:10.1038/nri3279. PMC 7097784. PMID 22918468.
- ^ a b Glusman G, Rowen L, Lee I, Boysen C, Roach JC, Smit AF, et al. (September 2001). "Comparative genomics of the human and mouse T cell receptor loci". Immunity. 15 (3): 337–49. doi:10.1016/s1074-7613(01)00200-x. PMID 11567625.
- ^ Deakin JE, Parra ZE, Graves JA, Miller RD (2006). "Physical mapping of T cell receptor loci (TRA@, TRB@, TRD@ and TRG@) in the opossum (Monodelphis domestica)". Cytogenetic and Genome Research. 112 (3–4): 342K. doi:10.1159/000089901. PMID 16484802.
- ^ a b Dushek O, Goyette J, van der Merwe PA (November 2012). "Non-catalytic tyrosine-phosphorylated receptors". Immunological Reviews. 250 (1): 258–76. doi:10.1111/imr.12008. PMID 23046135. S2CID 1549902.
- ^ Allison JP, McIntyre BW, Bloch D (November 1982). "Tumor-specific antigen of murine T-lymphoma defined with monoclonal antibody". Journal of Immunology. 129 (5): 2293–2300. doi:10.4049/jimmunol.129.5.2293. PMID 6181166. S2CID 13249566.
- ^ Meuer SC, Fitzgerald KA, Hussey RE, Hodgdon JC, Schlossman SF, Reinherz EL (February 1983). "Clonotypic structures involved in antigen-specific human T cell function. Relationship to the T3 molecular complex". The Journal of Experimental Medicine. 157 (2): 705–719. doi:10.1084/jem.157.2.705. PMC 2186929. PMID 6185617.
- ^ Haskins K, Kubo R, White J, Pigeon M, Kappler J, Marrack P (April 1983). "The major histocompatibility complex-restricted antigen receptor on T cells. I. Isolation with a monoclonal antibody". The Journal of Experimental Medicine. 157 (4): 1149–1169. doi:10.1084/jem.157.4.1149. PMC 2186983. PMID 6601175.
- ^ Yanagi Y, Yoshikai Y, Leggett K, Clark SP, Aleksander I, Mak TW (8 March 1984). "A human T cell-specific cDNA clone encodes a protein having extensive homology to immunoglobulin chains". Nature. 308 (5955): 145–149. Bibcode:1984Natur.308..145Y. doi:10.1038/308145a0. PMID 6336315. S2CID 4229210.
- ^ Hedrick SM, Cohen DI, Nielsen EA, Davis MM (8 March 1984). "Isolation of cDNA clones encoding T cell-specific membrane-associated proteins". Nature. 308 (5955): 149–153. Bibcode:1984Natur.308..149H. doi:10.1038/308149a0. PMID 6199676. S2CID 4273688.
- ^ Janeway Jr CA, Travers P, Walport M, et al. (2001). Immunobiology: The Immune System in Health and Disease. 5th edition. Glossary: Garland Science.
- ^ Kieke MC, Shusta EV, Boder ET, Teyton L, Wittrup KD, Kranz DM (May 1999). "Selection of functional T cell receptor mutants from a yeast surface-display library". Proceedings of the National Academy of Sciences of the United States of America. 96 (10): 5651–6. Bibcode:1999PNAS...96.5651K. doi:10.1073/pnas.96.10.5651. PMC 21915. PMID 10318939.
- ^ Sundberg EJ, Deng L, Mariuzza RA (August 2007). "TCR recognition of peptide/MHC class II complexes and superantigens". Seminars in Immunology. The Structure and Function of Antigen Receptors. 19 (4): 262–271. doi:10.1016/j.smim.2007.04.006. PMC 2949352. PMID 17560120.
- ^ Janeway CA, Travers P, Walport M, et al. (2001). "The Generation of Lymphocyte Antigen Receptors". Immunobiology: The Immune System in Health and Disease (5th ed.). Garland Science.
- ^ a b Call ME, Pyrdol J, Wiedmann M, Wucherpfennig KW (December 2002). "The organizing principle in the formation of the T cell receptor-CD3 complex". Cell. 111 (7): 967–79. doi:10.1016/s0092-8674(02)01194-7. PMC 3420808. PMID 12507424.
- ^ Smith-Garvin JE, Koretzky GA, Jordan MS (2009). "T cell activation". Annual Review of Immunology. 27: 591–619. doi:10.1146/annurev.immunol.021908.132706. PMC 2740335. PMID 19132916.
- ^ a b Feinerman O, Germain RN, Altan-Bonnet G (February 2008). "Quantitative challenges in understanding ligand discrimination by alphabeta T cells". Molecular Immunology. 45 (3): 619–31. doi:10.1016/j.molimm.2007.03.028. PMC 2131735. PMID 17825415.
- ^ Yang H, Buisson S, Bossi G, Wallace Z, Hancock G, So C, et al. (November 2016). "Elimination of Latently HIV-infected Cells from Antiretroviral Therapy-suppressed Subjects by Engineered Immune-mobilizing T-cell Receptors". Molecular Therapy. 24 (11): 1913–1925. doi:10.1038/mt.2016.114. PMC 5154472. PMID 27401039.
- ^ Blum JS, Wearsch PA, Cresswell P (2013). "Pathways of antigen processing". Annual Review of Immunology. 31: 443–73. doi:10.1146/annurev-immunol-032712-095910. PMC 4026165. PMID 23298205.
- ^ Evavold BD, Allen PM (May 1991). "Separation of IL-4 production from Th cell proliferation by an altered T cell receptor ligand". Science. 252 (5010): 1308–10. Bibcode:1991Sci...252.1308E. doi:10.1126/science.1833816. PMID 1833816.
- ^ Kersh GJ, Allen PM (October 1996). "Structural basis for T cell recognition of altered peptide ligands: a single T cell receptor can productively recognize a large continuum of related ligands". The Journal of Experimental Medicine. 184 (4): 1259–68. doi:10.1084/jem.184.4.1259. PMC 2192852. PMID 8879197.
- ^ Donermeyer DL, Weber KS, Kranz DM, Allen PM (November 2006). "The study of high-affinity TCRs reveals duality in T cell recognition of antigen: specificity and degeneracy". Journal of Immunology. 177 (10): 6911–9. doi:10.4049/jimmunol.177.10.6911. PMID 17082606.
- ^ Cole DK, Pumphrey NJ, Boulter JM, Sami M, Bell JI, Gostick E, et al. (May 2007). "Human TCR-binding affinity is governed by MHC class restriction". Journal of Immunology. 178 (9): 5727–34. doi:10.4049/jimmunol.178.9.5727. PMID 17442956.
- ^ Whitty A, Raskin N, Olson DL, Borysenko CW, Ambrose CM, Benjamin CD, Burkly LC (October 1998). "Interaction affinity between cytokine receptor components on the cell surface". Proceedings of the National Academy of Sciences of the United States of America. 95 (22): 13165–70. Bibcode:1998PNAS...9513165W. doi:10.1073/pnas.95.22.13165. PMC 23746. PMID 9789059.
- ^ a b Altan-Bonnet G, Germain RN (November 2005). "Modeling T cell antigen discrimination based on feedback control of digital ERK responses". PLOS Biology. 3 (11): e356. doi:10.1371/journal.pbio.0030356. PMC 1262625. PMID 16231973.
- ^ a b c Dushek O, Aleksic M, Wheeler RJ, Zhang H, Cordoba SP, Peng YC, et al. (June 2011). "Antigen potency and maximal efficacy reveal a mechanism of efficient T cell activation". Science Signaling. 4 (176): ra39. doi:10.1126/scisignal.2001430. PMC 4143974. PMID 21653229.
- ^ Huang J, Brameshuber M, Zeng X, Xie J, Li QJ, Chien YH, et al. (November 2013). "A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells". Immunity. 39 (5): 846–57. doi:10.1016/j.immuni.2013.08.036. PMC 3846396. PMID 24120362.
- ^ Miller MJ, Hejazi AS, Wei SH, Cahalan MD, Parker I (January 2004). "T cell repertoire scanning is promoted by dynamic dendritic cell behavior and random T cell motility in the lymph node". Proceedings of the National Academy of Sciences of the United States of America. 101 (4): 998–1003. Bibcode:2004PNAS..101..998M. doi:10.1073/pnas.0306407101. PMC 327133. PMID 14722354.
- ^ McKeithan TW (May 1995). "Kinetic proofreading in T-cell receptor signal transduction". Proceedings of the National Academy of Sciences of the United States of America. 92 (11): 5042–6. Bibcode:1995PNAS...92.5042M. doi:10.1073/pnas.92.11.5042. PMC 41844. PMID 7761445.
- ^ Dushek O, van der Merwe PA (April 2014). "An induced rebinding model of antigen discrimination". Trends in Immunology. 35 (4): 153–8. doi:10.1016/j.it.2014.02.002. PMC 3989030. PMID 24636916.
- ^ Lever M, Maini PK, van der Merwe PA, Dushek O (September 2014). "Phenotypic models of T cell activation". Nature Reviews. Immunology. 14 (9): 619–29. doi:10.1038/nri3728. PMID 25145757. S2CID 14274400.
- ^ von Essen MR, Kongsbak M, Geisler C (2012). "Mechanisms behind functional avidity maturation in T cells". Clinical & Developmental Immunology. 2012: 163453. doi:10.1155/2012/163453. PMC 3351025. PMID 22611418.
- ^ a b c d e f g h Murphy KM, Weaver C (22 March 2016). Janeway's immunobiology (Ninth ed.). W.W. Norton & Company. ISBN 978-0815345510.
- ^ a b van der Merwe PA, Dushek O (2011). "Mechanisms for T cell receptor triggering". Nature Reviews Immunology. 11 (1): 47–55. doi:10.1038/nri2887. PMID 21127503. S2CID 22423010.
- ^ Abram CL, Lowell CA (March 2007). "The expanding role for ITAM-based signaling pathways in immune cells". Science's STKE. 2007 (377): re2. doi:10.1126/stke.3772007re2. PMID 17356173. S2CID 44314604.
- ^ Nika K, Soldani C, Salek M, Paster W, Gray A, Etzensperger R, et al. (June 2010). "Constitutively active Lck kinase in T cells drives antigen receptor signal transduction". Immunity. 32 (6): 766–77. doi:10.1016/j.immuni.2010.05.011. PMC 2996607. PMID 20541955.
- ^ Tang Q, Subudhi SK, Henriksen KJ, Long CG, Vives F, Bluestone JA (May 2002). "The Src family kinase Fyn mediates signals induced by TCR antagonists". Journal of Immunology. 168 (9): 4480–7. doi:10.4049/jimmunol.168.9.4480. PMID 11970992.
- ^ Salmond RJ, Filby A, Qureshi I, Caserta S, Zamoyska R (March 2009). "T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance". Immunological Reviews. 228 (1): 9–22. doi:10.1111/j.1600-065X.2008.00745.x. PMID 19290918. S2CID 46343285.
- ^ a b c d e f Huse M (May 2009). "The T-cell-receptor signaling network". Journal of Cell Science. 122 (Pt 9): 1269–73. doi:10.1242/jcs.042762. PMID 19386893.
- ^ "UniProtKB - P06239 (LCK_HUMAN)". Uniprot. Retrieved 7 May 2020.
- ^ Essen LO, Perisic O, Katan M, Wu Y, Roberts MF, Williams RL (February 1997). "Structural mapping of the catalytic mechanism for a mammalian phosphoinositide-specific phospholipase C". Biochemistry. 36 (7): 1704–18. doi:10.1021/bi962512p. PMID 9048554.
External links
[edit]- T-Cell+Receptor at the U.S. National Library of Medicine Medical Subject Headings (MeSH)